

PDMSN - July 3, 2007 Full Issue
Vol. 1 No. 2
Off-Label Device Marketing Under Increasing Scrutiny
The marketing of products — particularly medical devices — for purposes for which they were not developed, tested or intended is coming under increased scrutiny by the FDA and Congress.
Increasing federal and state healthcare costs, spillover from off-label violations in the pharmaceutical arena and controversy surrounding the use of drug eluting stents (DESs) have focused attention on the use of language in device advertising to promote uses not intended for the target population. This also comes at a time when the Democratic-controlled Congress is champing at the bit to investigate medical marketing practices after 12 years of being out of power.
For example, in December 2006, the FDA’s Circulatory System Devices Panel Meeting found that a majority of DESs “are implanted in patients or vessels with characteristics different than those studied to support marketing approval.” The panel generally agreed that off-label use of DESs is associated with an increased risk of stent thrombosis, death and myocardial infarction as compared to on-label use.
Later, at a March 12 meeting on off-label promotion of biliary stents, it was noted that up to 90 percent of the market consists of sales to catheter labs where these stents are used in vascular procedures. The problem is that they’re indicated for propping open the ducts that carry digestive fluids in patients with end-stage pancreatic cancer. The FDA called in more than a dozen manufacturers and took them to task for promoting these stents at cardiovascular conferences and listing them in the cardiovascular sections of catalogs. The agency gave these companies three weeks to change these practices.
FDA’s Purview
The FDA can’t legally regulate the practice of medicine, but it can regulate manufacturers and their marketing practices. If the agency sees that the majority of a product’s use is off-label, it may suspect that the manufacturer is using off-label promotional practices.
The Office of the Inspector General (OIG) generally addresses these concerns in the context of lawsuits brought by whistle-blowers — sales reps or members of the target audience — under the False Claims Act. Under the act, which applies to Medicaid reimbursement, each violation is subject to a fine of $5,000 to $11,000, and the violator is subject to triple damages. Whistle-blowers can receive 15 to 30 percent of the damages. Companies in violation are subject to exclusion from federal programs, which in itself can be a crippling punishment.
Civil and Criminal Penalties
Increasingly, courts and regulators are signaling that they’re ready to bear down hard on off-label device marketing. Consider:
- Assistant U.S. Attorney James Sheehan from the Eastern District of Pennsylvania, who was recently nominated as New York State’s Medicaid Inspector General, has stated along with other state officials that his enforcement efforts will encompass off-label marketing of medical devices;
- In the Caputo case in 2006, the U.S. District Court for the Northern District of Illinois sentenced the compliance officer of medical device manufacturer AbTox to six years of imprisonment after he was convicted for inter alia off-label device manufacturing; and
- In late March, Rep. Henry Waxman, (D-Calif), chairman of the House Committee on Oversight and Government Reform, wrote to five companies — including two device manufacturers — requesting copies of all internal and external materials and plans related to marketing stents for potential off-label use.
Existing Guidelines Sketchy
Don’t look to existing guidelines for black and white rules on off-label marketing. Many of the guidelines focus on such issues as what constitutes improper remuneration between manufacturers and healthcare professionals. AdvaMed’s latest Code of Ethics on Interactions with Healthcare Professionals has no mention of off-label promotions, instead highlighting issues such as gifts and travel reimbursement. A March 2007 PricewaterhouseCoopers study found that “the appropriate use of data from postmarket studies without violating off-label promotion rules” was a gray area in the current code.
The OIG’s April 2003 voluntary guidance for pharmaceutical companies — which applies to device manufacturers as well — emphasizes anti-kickback provisions but is silent on off-label promotions.
Helpful Hints
In the absence of clear-cut guidance, some good rules of thumb include:
- Limit sales presentations and other marketing materials to approved indications for a device;
- Avoid discussing off-label uses in any marketing materials; and
- If there is a need to mention off-label use — for example, in reference to an ongoing clinical trial — make sure to state prominently that the product has NOT been approved for that indication.
It’s permissible to distribute journal articles that discuss off-label use in some circumstances, but be sure the information is from a credible, objective source, that the entire article is distributed, rather than excerpts, and that it contains a disclaimer that this use is not FDA-approved. The request for the article must be initiated by a healthcare professional, and the company should keep records of such requests and distributions.
The bottom line is that in the current environment, it is a much safer bet to simply avoid distributing such material. There are other ways for physicians to obtain clinical information that will not put your company (or even your person) at risk for prosecution. — Todd Clark
Detailing Dominates Pharma Marketing Spending
Detailing continues to account for the lion’s share of drug industry dollars when it comes to marketing drugs, a new study by Verispan shows.
The 2006 Year in Review by Verispan, a provider of healthcare data and analysis, examines trends in various kinds of marketing used by the pharmaceutical industry. It concludes that detailing is still the backbone of drug promotion, accounting for 48 percent of all pharmaceutical marketing dollars.
Spending on the office-based sales force grew by 14 percent from 2003 to 2006, but as a percentage of combined promotional activity, detailing’s share fell from 46 percent to 42 percent.
Promotional Allocations
The pharmaceutical industry invested $19.7 billion in promotional activities in 2006, an increase of 3 percent over 2005, but nowhere near the 18 percent and 20 percent posted for 2003 and 2004, respectively. Detailing in doctors’ offices accounted for the largest share — 42 percent — of promotional activity. Detailing in hospitals accounted for 6 percent. Together that accounts for nearly half of all promotional spending.
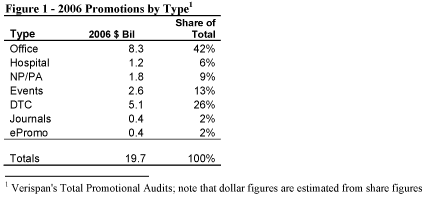
Direct-to-consumer (DTC) advertising accounted for the next largest share of dollars, or 26 percent. Trends in DTC and other forms of promotion will be examined in more detail in the next issue of the PDMSN Advisor.
Despite detailing’s foothold over pharmaceutical promotional dollars, a steady reduction in sales representatives has resulted in a 5.9 percent decline in sales calls, from a peak of 97.3 million in 2004 to 91.6 million in 2006. Headcounts have been declining since 2005, when the number of sales reps in the top 40 pharmaceutical companies dropped by 0.1 percent. It fell an additional 2 percent as the result of more substantial layoffs in 2006.
Less Bang for the Buck
Looking more closely, the data show that the number of actual detailing visits fell faster (by 5.9 percent) than the number of reps (by 2 percent) over the past two years. In a nutshell, that means the reps still out there are less productive. The average rep made about 917 calls per year in 2006, compared with 954 in 2004, which translates into a productivity decline of 3.8 percent. Apparently, not only are pharmaceutical companies cutting their sales forces, but they appear to be getting less out of their reps.
It is too early to draw sweeping conclusions from this. One of the driving factors may be that the industry has moved more toward specialty products such as biologics and oncology drugs. Relative to the big primary care blockbusters, these categories have smaller target audiences and thus would require fewer calls per rep (there are approximately 8,000 oncologists versus more than 50,000 internists). A less benign explanation is growing evidence that physicians are limiting or shutting out access to reps.
Decline in Journal Advertising
Medical journal advertising continues its slow fade. After languishing for over a decade, the $400 million in total spending on medical journal ads in 2006 was almost 20 percent lower than in 1996. It is not a coincidence that 1996 was the last year before the industry embraced DTC in a big way.
Ten years ago, it would have been unthinkable for major product producers to ignore journals entirely, but that is common practice today. For example, many mass market blockbuster products have not introduced new journal ads for five years or more. Journal publishers have long hoped DTC would hit a saturation point that would channel marketing funds back to them, but they’re still waiting.
Journals and other forms of marketing could be the beneficiaries if Congress heeds the growing demand for a ban on DTC during the first year or two of product availability. But publishers beware: even if such a ban gets passed, journals may not be the first place the funds get redirected. — Todd Clark
Lawmakers Discuss Creating National Gift Disclosure Laws for Physicians
Sen. Herb Kohl (D-Wis.) proposed creating a national registry to disclose gifts physicians receive from the pharmaceutical industry during a Senate Special Committee on Aging hearing.
Several witnesses voiced their support for the registry during the June 27 hearing, but warned that it would not be a panacea for conflicts of interest. The real problem lies with the conflicts, not with disclosing the conflicts, National Legislative Association on Prescription Drug Prices Executive Director Sharon Treat said. “Transparency alone may not be enough,” she added.
There is a trend of physicians changing their prescribing habits after meeting with industry representatives, Deputy Director of Public Citizen’s Health Research Group Peter Lurie said. Physicians make prescription decisions based on marketing, not on patient safety, he said.
According to a recent study in the New England Journal of Medicine, 94 percent of physicians have received food and beverages, medication samples and other gifts from drug companies, Kohl said. However, physicians and academic medical centers have been slow to respond to the issue, and “progress in extricating medicine from industry influence has been minimal,” Tufts University School of Medicine professor Jerome Kassirer said.
Any national laws to enhance transparency and disclosure of industry gifts should not preempt state laws, which could contain stronger restrictions, Lurie added.
“States are passing laws because there is a regulatory and enforcement void,” Treat said. Although the state laws may not be perfect, they are still addressing issues not covered by the federal government, she added. As of this month, at least 30 states had introduced or enacted laws on topics such as disclosing marketing spending, protecting patient privacy and restricting drug advertising, according to Treat.
However, none of the existing state statutes require device or biologic manufacturers to report payments to physicians, according to Lurie. A national registry for disclosing physician gifts should apply to all medical products, he said.
Lurie called upon the FDA, the FTC and state governments to enforce existing state laws. Kassirer asked Congress to fund an Institute of Medicine study on the amount and kinds of gifts industry representatives give to physicians.
Representatives from PhRMA and the American Medical Association (AMA) defended industry marketing practices and said their guidelines already regulated gift-giving to physicians.
The purpose of pharmaceutical promotion is to educate physicians, PhRMA Senior Assistant General Counsel Marjorie Powell said. PhRMA has a code that provides guidance on how industry sales representatives can maintain ethical relationships with healthcare professionals, she added.
In addition, the AMA continually revises its ethical guidelines governing physician interaction with industry members, AMA’s Council on Ethical and Judicial Affairs Chair Robert Sade said. For example, the guidelines indicate that a gift should mainly benefit patients and that a gift cannot be given in exchange for any other action, such as prescribing a certain drug.
State legislators who tried to write bills with gift disclosure laws have not “fully understood the complexities of what they were dealing with,” Powell said, adding that there would need to be much discussion before creating a national disclosure law.
Powell and Sade said there had been a change in industry behavior since PhRMA instituted its voluntary guidelines in 2002, although public perception has not yet changed.
However, Sen. Claire McCaskill (D-Mo.) disagreed, saying, “Your industry has got its head in the sand if you think you’ve turned a corner on this.”
“There’s more than a perception — there’s an issue, there’s a problem,” Kohl agreed. He said he would be working with Sen. Chuck Grassley (R-Iowa) and others to create “something concrete” for legislation creating new disclosure laws. — Emily Ethridge
Growing Asian Healthcare Market Expands Opportunities for Devicemakers
Per capita healthcare spending in Asia is expected to increase by almost 64 percent by 2010, expanding markets for medical devicemakers, according to a new report from Kalorama Information.
The report, “Asian Medical Device Markets,” says the market in Asia has experienced “constant growth” over the last decade, despite evolving regulatory and technological factors.
For the 13 countries in Asia, the report said the combined healthcare expenditure was $753 billion in 2005. But the report specifically focused on the healthcare sectors in five countries — Australia, China, India, Japan and South Korea — which Kalorama said have a combined value of approximately $31.63 billion in 2007.
By 2010, these five key markets are expected to total $43.08 billion, with China, Australia and India showing “exceptional growth.”
These markets could present important opportunities for devicemakers as the medical device markets of all five countries have “very large import components.”
In fact, for Australia and India, imports make up as much as 90 percent of the medical device market. Similarly, imports, primarily from the U.S. and Europe, make up almost 70 percent of the medical device market in China, the report said.
Key Markets
The domestic Australian medical device market was estimated to be worth approximately $2 billion in 2006 and is expected to grow at a compound annual growth rate (CAGR) of roughly 14.57 percent until 2010.
One of the key characteristics of the Australian medical device industry is that most medical devices produced are exported, while most medical devices consumed are imported, Kalorama said.
Another of the three major growth markets is China, estimated to be worth roughly $5.03 billion in 2006. Imports make up almost 70 percent of this market, which is expected to grow at a CAGR of almost 24 percent until 2010, the report said.
Technologies such as medical imaging, ultrasound probe technology and fiber endoscope-related technology are being introduced in China to satisfy increasing demand for products that meet international standards.
But industry growth is inhibited by doctors’ limited knowledge about high-technology devices, the “very small” demand for refurbished items and the “fragmented and unregulated” nature of the market, according to Kalorama.
India also has a “highly unregulated regulatory setup,” but is in the process of establishing regulatory norms, the report said. The medical device market there increased from approximately $550 million in 1993 to roughly $1.3 billion in 2006 and is expected to grow at a rate of roughly 7.2 percent annually until 2010.
Manufacturers may want to focus on India’s private sector, which held 80 percent of the healthcare delivery market in 2006 and “is expected to drive the development of infrastructure in the healthcare industry” over the next several years.
The Japanese medical device market, worth $19.27 billion in 2006, is the largest in the region and the second largest in the world, Kalorama said. However, it is expected to show the least growth — at a CAGR of 1.49 percent over the next five-year period — among the five countries.
Demand for Cutting Edge Technology
The demand for imported medical devices is still expected to increase, along with the demand for devices with “cutting-edge technology” manufactured by U.S. companies. In addition, because of its aging population and shortage of hospital beds, high-quality homecare equipment has a potential market in the country.
However, the saturation level of the market and the “rigid laws and regulations” of the country are limiting market growth.
Finally, the South Korean medical device industry is estimated to have been $2.61 billion in 2006, with an expected CAGR of 5.1 percent until 2010, to reach roughly $3.18 billion.
Favorable government policies and R&D support have helped the industry, the report said. The demand for homecare medical devices and self-treatment devices is growing and imports have been a significant component of the South Korean medical device market in recent years.
However, increasing medical expenses and strict requirements for market entry are the main growth inhibitors for the market. — April Astor
Misleading Drug Ads on the Rise, FDA Officials Say
Pharmaceutical advertising increasingly includes unsubstantiated claims about the safety and efficacy of drug products while downplaying or even failing to completely discuss the risks associated with taking them, Thomas Abrams, the director of the FDA’s Division of Drug Marketing, Advertising and Communication (DDMAC), said.
As a result, DDMAC has sent unusually large numbers of warning letters to pharmaceutical companies regarding these and related issues — 15 in 2005, 14 in 2006 and six in the first five months of 2007, compared with an average of four or five warning letters a year in previous years, Abrams said. His remarks came June 19 at the Drug Information Association’s 43rd Annual Meeting in Atlanta.
“We’re hearing from the industry about increased competition, causing marketing to want to ‘push the envelope,’” Abrams said. As well as causing the FDA to take enforcement actions, drug advertising that overplays safety and efficacy while underplaying risks can also “tarnish the industry” over time, he added.
The surge in warning letters from DDMAC is also due to the division’s prioritizing its work load “to get to the most serious violations first,” Abrams said.
The Advertising and Promotional Labeling Branch (APLB) at CBER had a surge in enforcement actions in 2004 that has tailed off since then, but many of the issues are the same as those DDMAC has been dealing with, APLB Chief Ele Ibarra-Pratt said. “If you have to squint to read the risk information but not the efficacy information [on a biologic product label], it’s a problem,” she said.
APLB places a priority on reviewing direct-to-consumer advertising, Ibarra-Pratt added.
Other problems in drug advertising that DDMAC and APLB have recently found include promotion of products for broader indications than those for which they are approved, and unsubstantiated comparative or superiority claims, Abrams and Ibarra-Pratt said.
DDMAC has seen the “pushing the envelope” phenomenon “in a few cases, not many,” Abrams said, stressing that the problems he was talking about affect a minority of drug advertising.
Abrams and Ibarra-Pratt also discussed ongoing changes that impact their offices. For example, if the user fee legislation now before Congress becomes law, DDMAC will be expanding to take on the new work, Abrams said. If the law passes, a company that submits 150 proposed television ads for review in a year would pay approximately $42,000 per ad, he said.
Companies submitting applications to APLB should be aware that the FDA’s esubmission gateway has been up since August 2006, and that the new physician’s labeling rule for human prescription drug and biological products has been in effect since June 30, 2006, Ibarra-Pratt said. Her branch is still receiving submissions under the old format and these are no longer valid, she said. — Martin Gidron
Generic Industry Experiencing a ‘Golden Age’
An aging population, increasingly consumer-directed healthcare, Medicare Part D and a surge in generic drug availability in key therapeutic areas are the main drivers of growth in the generic market, according to healthcare industry experts.
Generic drugs accounted for 60 percent of total drugs dispensed in 2006, and further growth predicted in 2007 and 2008 make this the “golden age” of generics, Ed Pezalla, vice president and medical director of Prescription Solutions, said at the Pharmaceutical Care Management Association’s Pharmacy Benefit Management & Generic Pharmaceutical Issues Symposium last month.
Medco Chairman and CEO David Snow, who gave the keynote address at the conference, said that more than $62 billion worth of brand drugs are expected to go off patent within the next five years, presenting boundless generic product growth opportunity.
In 2006, new generic drug availability in three important categories — statins, antidepressants and inhaled nasal steroids — resulted in 22 percent growth in generic sales in these classes, Brian Sweet, chief clinical pharmacy officer at WellPoint, said at the conference. Top sellers last year included Teva Pharmaceutical’s generic Zocor (simvastatin) with $911 million and Apotex’s generic Plavix (clopidogrel bisulfate) with $902 million, he added.
This year, generic prescription volume, which increased more than 11 percent from April 2006 to April 2007, is driving overall prescription volume growth, Sweet said.
Key drugs that have gone generic so far this year have been Norvasc (amlodipine besylate) and Ambien (zolpidem tartrate), which had $4.7 billion and $2.8 billion in sales last year, Sweet said. Final approval of generic Zyrtec (cetirizine HCl), which had sales of $1 billion last year, is expected later in 2007, he added.
The market entry this year of generic Ambien and Norvasc will continue to drive generic prescription and sales growth in the future, Sweet said. He noted that generics will have greater influence on the drug market in 2007 than any other year as the market realizes the full impact of the $14 billion in branded drugs that were subject to generic competition in 2006.
In 2008, several more important drugs have the potential to go generic, including Fosamax (alendronate sodium), which had sales of $1.4 million in 2006. Pezalla predicted that when osteoporosis drugs such as Fosamax go generic, there is bound to be an upsurge in prescriptions because patients are probably foregoing treatment with the expensive brand drugs. “I think there are a lot of unfilled prescriptions,” he said.
The two experts also addressed how follow-on biologics might affect the generic market in the future. The Senate Health, Education, Labor and Pensions Committee recently passed the Biologics Price Competition and Innovation Act, S. 1695, which would allow the FDA to approve products as biosimilar to or interchangeable with brand biologics. The bill’s authors, including Sen. Edward Kennedy (D-Mass.), plan to attach the bill to the Prescription Drug User Fee Act reauthorization bill when the Senate and House meet to negotiate the final version later this month.
While generic companies are able to quickly manufacture and ship vast quantities of small-molecule generic drugs as soon as they are approved, it may not be so easy with biotech products. Nationwide availability of follow-on biologics might be “spotty,” Pezalla said, and unless products are approved as interchangeable, patients would not be able to switch back and forth between the brand and the low-cost follow-on.
Even at pharmacies where the follow-on products are available, patients are likely to be less trustworthy of biosimilar versions of life-sustaining cancer or diabetes treatments, so manufacturers and health plans may have to launch educational campaigns, Pezalla continued.
Snow, during his keynote address, called for support of follow-on biologics legislation, which he said would not only spur growth in the generic industry, but would promote new developments among innovator companies. — Breda Lund
PhRMA Slams Indian Price Controls, IP Failures
In its recent “2007 National Trade Estimate Report for India,” PhRMA objected to India’s plans to impose price controls on 354 more drugs, in addition to the 74 that currently have price controls. The trade organization also highlighted what it sees as intellectual property (IP) protection weaknesses and other trade barriers confronting the drug industry in India.
PhRMA expressed concern about the Indian government’s proposed requirement for mandatory price negotiations as a condition of marketing approval for patented drugs launched in India after Jan. 1, 2005. “PhRMA members feel that this proposal represents an effort to effectively nullify the benefits of product patent protection, as there is no reference to the intellectual property intrinsic in the products. This appears to be a backdoor effort to circumvent India’s WTO [World Trade Organization] commitment to provide product patent protection for patented and imported medicines.”
“This government pricing regime, combined with the lack of an effective patent or other intellectual property protection, makes the commercial environment in India unfavorable for research-based companies,” PhRMA added.
Other issues of concern for PhRMA regarding the Indian Patents Act include:
- Scope of patentability;
- Compulsory licensing provisions;
- Lack of protection for mailbox application patents;
- Misuse of the pre-grant opposition processes;
- A lack of effective measures to prevent unfair commercial use of data;
- The Indian government’s failure to adopt standards for patentability to bring the country into the mainstream of patent practices; and
- The need to deal with issues relating to compulsory licensing for pharmaceutical products that fall short of India’s WTO obligations. At present, Indian compulsory licensing “inappropriately erode[s] the rights of the patent holder,” PhRMA said.
“These issues bring India into conflict with its minimum international obligations,” PhRMA said. “Almost a year and half after passage of the new product patent regime, India has issued only one patent for a pharmaceutical product.” — Martin Gidron
Proposed Patent Reforms Worry Device Industry
Industry groups say the Patent Reform Act of 2007 threatens to “devastate life sciences investment” by weakening the patent system for medical devicemakers. Provisions regarding apportionment of damages in lawsuits, rulemaking authority for the Patent and Trademark Office (PTO) and post-grant patent oppositions will “create a chilling effect on the industry,” according to the Medical Device Manufacturers Association (MDMA).
The legislation passed the House Subcommittee on Courts, the Internet and Intellectual Property last month and is now pending markup by the full House Judiciary Committee. The Senate Judiciary Committee held a markup on its version of the legislation June 21.
Under the apportionment provision, damages for patent infringement would never be more than the economic value attributable to the innovation, AdvaMed said.
In the medical field, many “critically important” advances are incremental and build upon prior technology. By apportioning damages to the economic value attributable to the specific contributions over the prior art, the act would severely diminish the value of medical device patents, AdvaMed added.
MDMA agreed, saying, “Even if the patented invention is truly the reason for the sale of an entire product, but … is virtually costless to add to the product, the patentee would obtain virtually no royalty.”
Industry Opposes ‘Second Window’
The industry groups also oppose the “second window” in the post-grant opposition provision. This is a second chance to challenge a patent after the first year if the petitioner receives a notice of infringement or can prove the patent causes significant economic harm.
The Senate Judiciary Committee passed an amendment June 21 revising the post-grant opposition provision so that a petitioner must have both a notice of infringement and proof of significant economic harm, instead of one or the other.
Unlike many other industries, the life cycle of medical devices is relatively short — often no longer than seven to 10 years. The ability to quickly capture, and then maintain, market share over this short period is crucial, AdvaMed said.
If a competitor were allowed, at any time, to commence a post-grant opposition proceeding, it could continue to infringe on the patent during the proceedings, which can be lengthy. This would “significantly limit” the already short exclusivity period for medical devices, AdvaMed said.
AdvaMed said it is particularly concerned that the combination of limiting damages and allowing a second window of post-grant oppositions could encourage “patent pirates,” including foreign companies, to copy the inventions of U.S. medical device companies. After an infringement action is brought, an opposition proceeding could delay any litigation, AdvaMed said, adding that following a finding of infringement, it is possible that only minimal compensation would be paid. “This perverse outcome must be avoided.”
AdvaMed said that recently proposed changes to the bill by Rep. Howard Berman (D-Calif.) and several other lawmakers do not resolve its concerns. “The very limited proposed bases under which a company could not utilize the ‘second window’ will not lessen the impact this portion of the act would have on medical device companies.”
PTO Authority
In addition, substantive rulemaking should not be delegated to the PTO, the groups said. MDMA said the PTO has “made it clear” it wants to limit continuation filings to reduce backlog, despite opposition.
The enhanced rulemaking authority in the House version would permit these limits, significantly reducing the ability of smaller life sciences companies to secure coverage for their inventions, it added.
In early stages, companies do not have the resources to file claims for every invention disclosed in patent applications and they use continuations to overcome this limitation. Also, because there is usually a “critical time window” for the applicant to demonstrate value to prospective investors, there is great pressure to accept quick issuance of narrower claims, MDMA said. The applicant uses the continuation process to then pursue broader protection.
The committee is scheduled to continue working on the legislation June 28. — April Astor, Breda Lund
Tips of the Week
Devices:
- The Supreme Court agreed on June 25 to hear Riegel v. Medtronic. The facts of the case, which dates back to 1996, are not all that important. However, all FDA regulated companies should be watching to see how the court addresses the issue of preemption. Under this doctrine, manufacturers of FDA-approved products are immune to liability lawsuits in state courts because the requirements of complying with federally (i.e. FDA) mandated regulations trumps states law. Without it in place, device and drug manufacturers would be subject to lawsuits under differing state laws. The Court will hear the case in October.
- A report from analysts at Canaccord Adams, “Orthopedics 2007 And Beyond: Innovation and Demographics Drive Industry Growth,” indicates that worldwide sales of orthopedic devices reached $24 billion last year. The category is expected to grow by 10 percent annually. Joint implants are the largest segment — accounting for 41 percent of the total market (products for the spine are next at 18 percent). The fastest growing segment is orthobiologics which saw 22 percent sales growth in 2006. A few companies dominate most of the larger sub-segments, with the top three manufacturers having 78 percent share of the hip implant market, 74 percent of knee replacements and 68 percent of spinal implants. Companies considering whether to enter these mature arenas should note the report’s conclusion that “market share shifts are glacial due to strong sales rep/surgeon relationships. In orthopedics the sales reps do more than sell; they provide service and support that hospital does not want to or is not able to. As a result, strong allegiances are built between the surgeon and sales rep, resulting in a resistance of surgeons to changing suppliers.”
Pharma:
- Given the publicity surrounding new cancer therapies which cost well over $20,000 per patient annually — and the much lamented death of the primary care blockbuster — it’s no surprise that specialty drugs are the growth engine in the U.S. pharmaceutical market. Medco’s 2007 Drug Trend Report provides a snapshot of just how much of a driving factor specialty drugs are. According to the report, specialty pharmacy spending increased 16.1 percent in 2006 versus an overall average of only 2.8 percent. The biggest categories within the specialty drug pie are rheumatoid arthritis (25 percent), multiple sclerosis (18 percent) and cancer (16 percent). With a 9 percent increase in utilization and a 27 percent increase in unit costs, cancer drugs were the biggest contributor to growth.
- Recently the governor of Vermont signed “An Act Relating to Increasing Transparency of Prescription Drug Pricing and Information.” Among other things, the law prohibits use, sale or transfer of prescriber-identifiable data “for commercial advantage.” Passage occurred despite a federal court ruling that a similar law in neighboring New Hampshire was unconstitutional on free speech grounds (New Hampshire is appealing that ruling). According to his spokesman, the governor did not consider the constitutionality of the law. However, the New Hampshire court ruling was taken into account by the legislature. They appear to have done a good job of tailoring the act so that it stands a good chance of prevailing against legal challenges. In contrast to the New Hampshire law, which included penalties of up to two years in jail, Vermont’s law calls for fines rather than imprisonment. The law allows physicians to opt-in to data sharing and there are a number of exceptions that were not included in the New Hampshire law, including a provision allowing for marketing use provided that it is not identifiable at the prescriber level. Vermonters spend approximately $524 million on prescription and OTC drugs — less than 0.2 percent of the U.S. pharmaceutical market — so the law’s importance arises less from the amounts directly at stake than from the very real possibility that other states or Congress will copy the approach.
|
|||||||||
|
|||||||||
Upcoming Events
-
25Apr
-
07May
-
14May
-
30May
